
Tra i pilastri della ricerca finanziata dal Piano nazionale di Sviluppo Rurale, prima con il progetto LATTeco e ora nel LATTeco2, c’è la biodiversità: l’obiettivo è misurarla e mettere in campo tutte le azioni per poterla mantenere il più elevata possibile, soprattutto nelle popolazioni sottoposte ad intensa selezione come la Frisona Italiana. La perdita di biodiversità è infatti collegata all’aumento di omozigosi provocato dall’azione di selezione conosciuto dai più con il nome di “consanguineità“.
La misura della consanguineità genomica
Nell’era della genomica è possibile studiare la variabilità genetica, o biodiversità, analizzando la sequenza delle basi del DNA ed identificando le differenze che caratterizzano individui e popolazioni. Attraverso lo studio delle differenze fra i marcatori nel tempo è possibile ricostruire i percorsi migratori delle specie nei secoli, e seguire le tracce delle azioni di domesticazione e differenziazione delle diverse razze e popolazioni all’interno di una stessa specie.
Lo si fa quantificando le aree in cui c’è assenza di variazione tra individui della stessa specie o popolazione (aree di omozigosi) e quelle in cui c’è variabilità (eterozigosi) e facendo confronti fra individui nel tempo e nello spazio geografico.
La vicinanza o la distanza genetica nelle popolazioni sottoposte a selezione si misura anche attraverso la somiglianza o la diversità fra individui: parentela e consanguineità aumentano quando si autorizza la riproduzione di solamente pochi maschi miglioratori e il processo si accentua quando si selezionano intensamente sia i padri che le madri delle future generazioni.
L’azione della selezione tende a fissare gli alleli favorevoli e, nelle aree del genoma dove stanno i geni legati alle caratteristiche peculiari di una razza che sono state selezionate nel tempo, si crea una maggiore omozigosi in cui sono state ereditate caratteristiche identiche sia dalle linee paterne che materne. È in queste zone che si crea quella che inglese viene chiamata “identity by descent“, ovvero uguaglianza per discendenza, legata al fatto di avere progenitori imparentati fra loro nella genealogia: la consanguineità.
Una delle analisi classiche della variabilità fatte utilizzando le informazioni raccolte sul genoma è che prevede l’analisi delle sequenze di omozigosi (ROH = Runs of Homozigosity) lungo il genoma, ovvero segmenti di DNA in cui si susseguono in maniera ininterrotta dei genotipi omozigoti. È così che si misura la consanguineità genomica.
L’analisi della distribuzione delle ROH e dei marcatori (o snps) che sono inclusi con elevata frequenza nelle ROH permette l’individuazione delle Selection Signatures, regioni del genoma dove si verifica un calo dell’eterozigosi legato.
alla fissazione, dovuta alla delezione, dell’allele più favorevole ad un carattere di interesse come ad esempio la produzione. Queste “signatures” possono variare da una razza all’altra ad esempio fra le razze fortemente selezionate per il latte a confronto con quelle selezionate per mantenere la duplice attitudine.
Tra i parametri più importanti delle ROH vanno ricordate la lunghezza e la loro frequenza all’interno della popolazione. ROH lunghe sono indice di inbreeding (inteso come accoppiamento fra animali parenti) recente nel tempo, mentre ROH più corte indicano fenomeni di inbreeding lontani nel tempo, legati ad ascendenti comuni molte generazioni indietro che progressivamente sono stati frammentati dalle ricombinazioni avvenute lungo il DNA e influenzate dalla selezione naturale che contrasta la consanguineità “cattiva”, quella che cioè nasconde geni difettosi che riducono la vitalità dei soggetti e/o la loro capacità di riprodursi con successo e di trasmettere il difetto alle generazioni future.
Lo studio presentato all’EAAP nel 2020
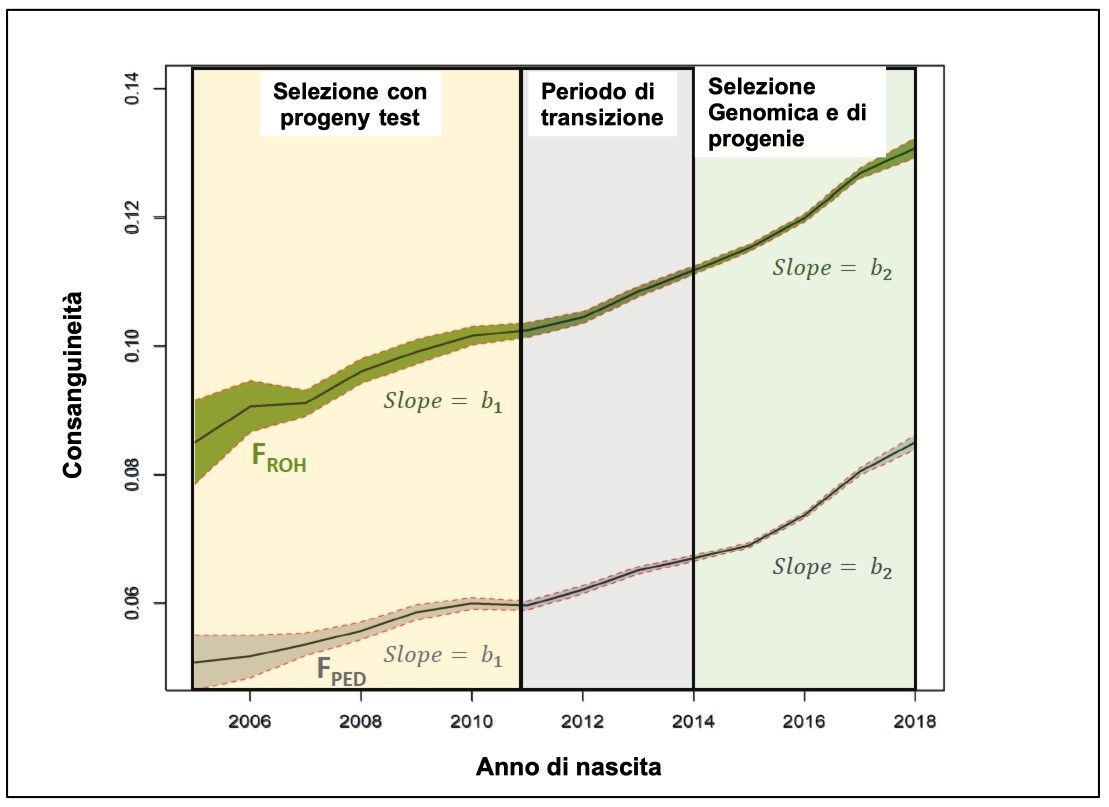
Sulla Frisona in Italia l’Università di Parma ha presentato al convegno della Società Europea di Zootecnia (EAAP) del 2020 i risultati di un primo lavoro di confronto fra la consanguineità calcolata da pedigree e quella stimata a partire dall’analisi delle RHO sulla popolazione femminile genotipizzata. La figura 1 riporta il confronto tra l’andamento della consanguineità basata su pedigree e RHO nel tempo, nel passaggio dalla selezione basata sulle prove di progenie a quella fondata sulla genomica. La scala delle consanguineità, tra due metodi di calcolo, è leggermente diversa ma l’andamento è molto simile. È evidente l’incremento della velocità di aumento di consanguineità nell’era della genomica. La correlazione fra le due misure è del 60%. Il cambio di “passo” tra “prove di progenie” e “genomica” è pari a +1,02 per la consanguineità misurata attraverso le RHO e di +3,02 quando osserviamo la differenza sulla scala dell’inbreeding basato sull’analisi del pedigree. I dati genomici sembrano dire che la consanguineità nel passato era già più elevata di quanto fosse possibile stimare attraverso le informazioni spesso “lacunose” del pedigree.
Figura 1 – Il confronto fra l’incremento di consanguineità calcolata in base al pedigree ed in base all’analisi genomica dell’omozigosi dal 2006 al 2018 nella popolazione Frisona genotipizzata (Fonte: modificato da Ablondi e coll., 2021, EAAP).
I lavori sono ancora in corso e puntano ad identificare lo strumento più appropriato per mettere a disposizione questa informazione più precisa e dettagliata a tutti gli allevatori che genotipizzano in modo che possa essere utilizzata dai piani di accoppiamento “genomici”, ad esempio.
Il progetto GENORIP
È tutt’ora in corso anche un progetto dell’Università di Milano che ha come obiettivo quello di mettere a punto uno strumento per l’identificazione dei riproduttori più fuori linea rispetto alla mandria che vede coinvolte oltre una decina di aziende. Il progetto si chiama GENORIP e prevede l’utilizzo di diversi approcci nel calcolo della consanguineità genomica per arrivare ad identificare quello più utile nella scelta dei tori da utilizzare in FA in grado di minimizzare la perdita di variabilità genetica nella mandria. L’obiettivo finale è dare a tutti uno strumento per lavorare sulla consanguineità prevista nella progenie in maniera più precisa e puntuale. Le aziende che hanno aderito al progetto hanno avuto la possibilità di genotipizzare tutta la mandria, di conoscere in maggiore dettaglio la composizione genetica e il livello di consanguineità delle bovine allevate, e di confrontarsi sul tema con i ricercatori dell’Università e fra di loro.
In sintesi
Sulla biodiversità e sulla consanguinetà la genomica offre la possibilità, ancora una volta, di mettere a punto strumenti più efficaci di monitoraggio e controllo. Si attende che vengano presto resi disponibili a tutti gli allevatori che già utilizzano la genomica per fare selezione in azienda.





Scrivi un commento
Devi accedere, per commentare.