
Sono molti gli allevatori e gli addetti ai lavori che utilizzano i risultati di una valutazione genetica o genomica. Lo fanno sfogliando cataloghi, scegliendo i tori ed accoppiandoli con le loro vacche. Chissà se si sono mai chiesti come si fa ad essere sicuri che questi risultati siano davvero i più corretti ed affidabili.
Introduzione
Wyatt Earp, uno sceriffo statunitense vissuto a cavallo tra il 1800 ed il 1900, è un nome forse non noto a tutti ma è una figura leggendaria perché ha ispirato tanti film western. Aveva anche l’innata capacità di riassumere in modo molto semplice aspetti piuttosto importanti ed infatti famoso è il suo detto: Fast is fine, but accuracy is everything – Essere veloci è importante, ma essere accurati lo è ancor di più. Ecco, questo motto, seppur nato in un altro ambito, si adatta perfettamente ai risultati di una valutazione genetica o genomica: è importante averli velocemente ma è ancora più importante che siano accurati.
Questo discorso è stato fatto tante volte ma in effetti la capacità di stimare indici accurati è uno dei punti cardine delle attività legate al miglioramento genetico. Le valutazioni genetiche sono un pò come le previsioni del tempo: cerco di prevedere oggi quello che succederà tra qualche giorno o settimana. Per farlo utilizzo le informazioni che ho raccolto sino ad ora e le conoscenze su quello che è successo nel tempo. Posso utilizzare modelli più o meno complessi ma alla fine a chi sta nel campo interessa sapere se pioverà davvero oppure no. Se le informazioni fornite saranno poco accurate la fiducia nel sistema verrà meno.
La necessità di essere però veloci ed accurati è aumentata negli ultimi anni, in conseguenza soprattutto dell’introduzione della genomica che ha portato sia all’utilizzo di modelli più complessi ma anche alla combinazione di informazioni diverse: tori selezionati solo sulla base delle informazioni delle figlie e tori selezionati solo sulla base delle informazioni genomiche. Un po’ come se le previsioni del tempo fossero fatte con dati raccolti dalle centraline tradizionali e da un certo punto in avanti anche dai droni. Queste due fonti di informazioni misurano la stessa cosa ma in modo leggermente diverso.
Da oltre 30 anni chi si occupa di miglioramento genetico ha sviluppato delle procedure per validare i risultati delle valutazioni genetiche (Boichard et al, 1995, Thompson R., 2001) e tra questi ce ne è una relativamente banale e che si basa sul cosiddetto troncamento dei dati. Questo metodo è oggi uno dei più utilizzati sia per la facilità di applicazione che per i risultati che produce. Vediamo quali parametri utilizza e come funziona.
Bias, accuratezza e dispersione
Le valutazioni genetiche utilizzano i dati raccolti sui singoli individui e le relazioni di parentela (tradizionali e genomiche) tra gli stessi per predire il valore genetico di ogni individuo presente in popolazione. Alcuni soggetti possono avere molti dati, altri possono non averli per niente ma alla fine tutti avranno un indice, indice che avrà una sua accuratezza che varierà in funzione della quantità di informazioni disponibili.
Le informazioni si aggiungono nel tempo e quindi il valore genetico potrebbe cambiare: quello che dico oggi potrebbe non essere vero domani. E questo sarebbe un problema. Dovrei invece essere sicuro che le informazioni fornite cambino poco. Citando Earp: devo dire cose accurate.
In pratica che cosa vuol dire? Vuol dire verificare che: 1. la differenza tra la stima fatta oggi e la stima fatta domani sia 0 o molto prossima allo 0, il cosiddetto bias; 2. la stima fatta con e senza dati fenotipici sia molto simile, la cosiddetta accuratezza; 3. la predizione fatta non sottostima e non sovrastima, la cosiddetta dispersione. Un modo per rispondere a queste domande è usare il cosiddetto troncamento dei dati, approccio utilizzato nel Metodo LR ((Legarra & Reverter, 2018).
Il metodo LR
Questo metodo di validazione, nato nel 1994 (Reverter et al, 1994) è tornato alla ribalta grazie alla sua semplicità di applicazione. Il concetto su cui si basa è sintetizzato nella figura 1.
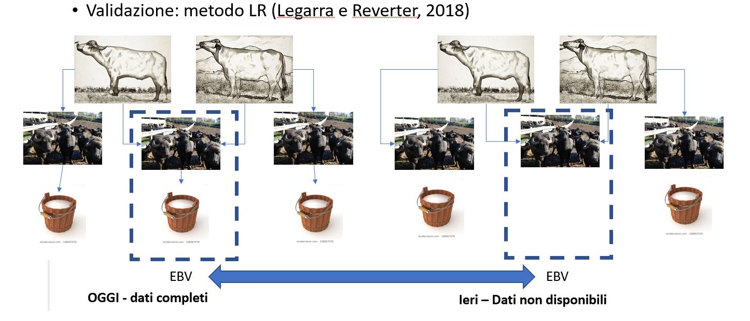
Figura 1. Validazione delle valutazioni genetiche/genomiche attraverso il cosiddetto troncamento dei dati. Si confrontano gli EBV di uno specifico gruppo di animali ottenuti con tutti i dati (parte sinistra) con quelli ottenuti con dati parziali o troncati, in cui gli stessi individui non hanno dati fenotipici (parte destra).
Il metodo si basa sulla stima degli indici in due momenti temporalmente diversi in modo tale che, per uno specifico gruppo di individui, gli indici vengano calcolati con tutte le informazioni disponibili (indice che ipotizziamo essere quello corretto perché stimato appunto con tutte le informazioni) oppure solo con una parte (solo con le parentele genomiche).
Ovviamente vorremmo che quest’ultimo sia il più vicino possibile a quello corretto. Per fare questo calcoliamo gli indici con i dati di oggi (parte sinistra della figura 1) e li consideriamo il gold standard. Poi identifichiamo un gruppo di individui per i quali vogliamo predire il valore genetico/genomico (ad esempio i tori giovani) e creiamo dei dati troncati (parte destra della figura 1). In questi dati troncati i soggetti di interesse non hanno dati fenotipici (se tori non hanno figlie o se femmine non hanno produzione), e quindi la stima del loro indice sarà basata solo sulle parentele genomiche.
A questo punto per ogni soggetto del gruppo di riferimento avrò due valori: quello che considero vero ottenuto con tutti i dati (gold standard) e quello parziale. In pratica sto simulando quello che succede normalmente: oggi stimo degli indici per dei soggetti che non hanno ancora dati.
Con i due indici posso quindi calcolare i 3 parametri di cui parlavo prima – bias, accuratezza e dispersione – e posso non solo confrontare tra loro modelli ed approcci diversi ma anche valutare l’impatto su gruppi diversi di animali (e.g. maschi o femmine).
Considerazioni finali
Il metodo LR è oggi uno dei più utilizzati proprio perché estremamente semplice e veloce da usare, sebbene si debba fare attenzione al gruppo di soggetti utilizzati per validarlo. Ad ogni modo un recente articolo pubblicato sul Journal of Dairy Science (Himmelbauer et al, 2023) ha dimostrato come questo metodo, considerando anche la sua semplicità di applicazione, fornisce i risultati migliori rispetto ad altri metodi disponibili, essendo in grado di identificare con successo la minor efficienza di un modello di valutazione genetica. Tra l’altro è facilmente applicabile a tutte le specie ed a tutti i tipi di caratteri (lineari o categorici) cosa non possibile per gli altri metodi attualmente disponibili. Wyatt Earp sarebbe soddisfatto: un metodo veloce ma soprattutto preciso.
Bibliografia Consultata
Boichard, D., Bonaiti, B., Barbat, A., & Mattalia, S. (1995). Three methods to validate the estimation of genetic trend for dairy cattle. Journal of dairy science, 78(2), 431-437.
Thompson, R. (2001). Statistical validation of genetic models. Livestock Production Science, 72(1-2), 129-134.
Reverter, A., Golden, B. L., Bourdon, R. M., & Brinks, J. S. (1994). Detection of bias in genetic predictions. Journal of animal science, 72(1), 34-37.
Legarra, A., & Reverter, A. (2018). Semi-parametric estimates of population accuracy and bias of predictions of breeding values and future phenotypes using the LR method. Genetics Selection Evolution, 50, 1-18.
Himmelbauer, J., Schwarzenbacher, H., Fuerst, C., & Fuerst-Waltl, B. (2023). Comparison of different validation methods for single-step genomic evaluations based on a simulated cattle population. Journal of Dairy Science, 106(12), 9026-9043.





Scrivi un commento
Devi accedere, per commentare.